Research Areas
Recent Research
“Genome-wide germline correlates of the epigenetic landscape of prostate cancer”
Houlahan KE, Shiah YJ, Gusev A, et al. Genome-wide germline correlates of the epigenetic landscape of prostate cancer. Nat Med. 2019;25(10):1615-1626. doi:10.1038/s41591-019-0579-z
View Publication
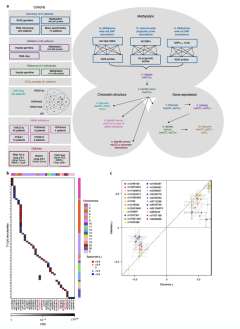
Oncogenesis is driven by germline, environmental and stochastic factors. It is unknown how these interact to produce the molecular phenotypes of tumors. We therefore quantified the influence of germline polymorphisms on the somatic epigenome of 589 localized prostate tumors. Predisposition risk loci influence a tumor's epigenome, uncovering a mechanism for cancer susceptibility. We identified and validated 1,178 loci associated with altered methylation in tumoral but not nonmalignant tissue. These tumor methylation quantitative trait loci influence chromatin structure, as well as RNA and protein abundance. One prominent tumor methylation quantitative trait locus is associated with AKT1 expression and is predictive of relapse after definitive local therapy in both discovery and validation cohorts. These data reveal intricate crosstalk between the germ line and the epigenome of primary tumors, which may help identify germline biomarkers of aggressive disease to aid patient triage and optimize the use of more invasive or expensive diagnostic assays.
“Widespread and Functional RNA Circularization in Localized Prostate Cancer”
Chen S, Huang V, Xu X, et al. Widespread and Functional RNA Circularization in Localized Prostate Cancer. Cell. 2019;176(4):831-843.e22. doi:10.1016/j.cell.2019.01.025
View Publication
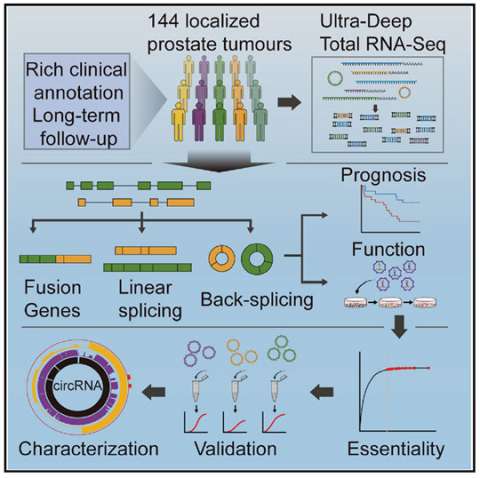
The cancer transcriptome is remarkably complex, including low-abundance transcripts, many not polyadenylated. To fully characterize the transcriptome of localized prostate cancer, we performed ultra-deep total RNA-seq on 144 tumors with rich clinical annotation. This revealed a linear transcriptomic subtype associated with the aggressive intraductal carcinoma sub-histology and a fusion profile that differentiates localized from metastatic disease. Analysis of back-splicing events showed widespread RNA circularization, with the average tumor expressing 7,232 circular RNAs (circRNAs). The degree of circRNA production was correlated to disease progression in multiple patient cohorts. Loss-of-function screening identified 11.3% of highly abundant circRNAs as essential for cell proliferation; for ∼90% of these, their parental linear transcripts were not essential. Individual circRNAs can have distinct functions, with circCSNK1G3 promoting cell growth by interacting with miR-181. These data advocate for adoption of ultra-deep RNA-seq without poly-A selection to interrogate both linear and circular transcriptomes.
“The Proteogenomic Landscape of Curable Prostate Cancer”
Sinha A, Huang V, Livingstone J, et al. The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell. 2019;35(3):414-427.e6. doi:10.1016/j.ccell.2019.02.005
View Publication
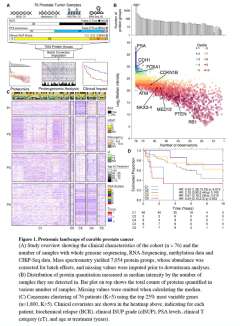
DNA sequencing has identified recurrent mutations that drive the aggressiveness of prostate cancers. Surprisingly, the influence of genomic, epigenomic, and transcriptomic dysregulation on the tumor proteome remains poorly understood. We profiled the genomes, epigenomes, transcriptomes, and proteomes of 76 localized, intermediate-risk prostate cancers. We discovered that the genomic subtypes of prostate cancer converge on five proteomic subtypes, with distinct clinical trajectories. ETS fusions, the most common alteration in prostate tumors, affect different genes and pathways in the proteome and transcriptome. Globally, mRNA abundance changes explain only ∼10% of protein abundance variability. As a result, prognostic biomarkers combining genomic or epigenomic features with proteomic ones significantly outperform biomarkers comprised of a single data type.