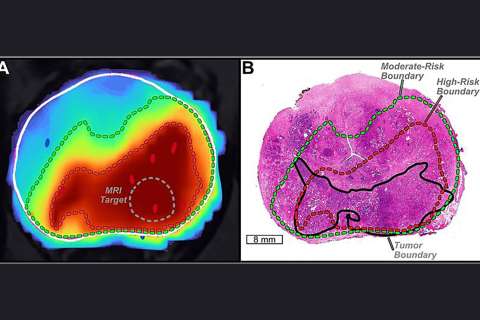
Like locking the door but opening windows, an immune checkpoint therapy temporarily slows glioblastoma (GBM) progression but fails to establish an effective anti-tumor microenvironment and appears to increase molecular interactions inhibiting long-term immune response, according to studies led by a UCLA research team.
In the Nov. 26 issue of Nature Communications, the researchers explain why PD-1 blockade – which is effective against several other cancers – is insufficient on its own against these deadly brain tumors. They also provide details on the immune landscape and propose future strategies and investigations.
“Primary brain tumors, such as glioblastoma, are remarkably resistant to immunotherapy. In preclinical studies, immunotherapy appears to be effective, and in a previous, small, phase 1 clinical trial using anti-PD-1 therapy before tumor-removal surgery, an interferon signature was induced and length of survival increased. But the treatment is not curative, suggesting there are other interactions within the microenvironment that this treatment does not address,” said lead author Robert M. Prins, PhD, a researcher at the UCLA Jonsson Comprehensive Cancer Center and a tumor immunologist and professor at the David Geffen School of Medicine at UCLA.
To identify immune-related mechanisms and interactions responsible for the limited success and ultimate failure of PD-1 blockade to stop brain tumor progression, Prins teamed up with Willy Hugo, PhD, and other colleagues from the UCLA Jonsson Comprehensive Cancer Center, several departments at UCLA Health, and the Parker Institute for Cancer Immunotherapy in San Francisco to analyze immune cell populations at the single-cell level with advanced single-cell technologies.
PD-1, a so-called “checkpoint protein,” helps T cells from the immune system properly regulate an attack, normally functioning as an “off switch” to protect normal cells. Many cancers, however, hijack the process to protect themselves from attack. In lung, skin and other cancers, the administration of monoclonal antibodies to inhibit the checkpoint protein enables T cells to resume attacking cancer cells.
In glioblastoma, the researchers found, PD-1 blockade initially increases the proportion and number of a subset of “progenitor exhausted” T cells and activates a population of conventional dendritic cells, but the immune microenvironment remains largely dominated by other immune cells (myeloid cells) that produce inflammatory molecules and are immunosuppressive. To be effective, PD-1 blockade may need to be combined with therapies targeting other checkpoint proteins and immunosuppressive pathways.
According to the authors, the results suggest that anti-PD-1 treatment initially activates T cells in the bloodstream, some of which migrate into the tumor microenvironment and produce interferon gamma.
Despite the positive early effects of anti-PD-1 treatment, a population of white blood cells (monocytes and macrophages) recruited into the tumor microenvironment also appears to respond to the interferon signaling in a maladaptive fashion. Anti-PD-1 therapy appears to increase interactions between these cells and two other checkpoints on T cells that downregulate immune responses, CTLA-4 and TIGIT.
These interactions may limit the magnitude and persistence of the anti-tumor T cell activities, Prins and Hugo said. The data suggest that anti-PD-1 treatment improves survival outcomes by increasing an anti-tumor T cell response, but the treatment also changes the tumor microenvironment and influences non-T cell populations. The effectiveness of the first response is curtailed by engagement of the additional T cell checkpoints, and new strategies that also target other immunosuppressive pathways may improve the strength and durability of the anti-tumor response.
Funding and acknowledgements:
This study was funded in part by the National Institutes of Health SPORE in Brain Cancer (P50CA211015) and NIH/NCI grant (1R01CA222695-01), NIH National Center for Advancing Translational Science - UCLA CTSI (UL1TR001881), the Parker Institute for Cancer Immunotherapy, the Brain Tumor Funder’s Collaborative, and the Cancer Research Institute.
Mass cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) Flow Cytometry Core Facility that is supported by NIH award P30 CA016042. The purchase of the Helios/CyTOF mass cytometer that was used in this work was, in part, supported by funds provided by the James B. Pendleton Charitable Trust. Single-cell RNA sequencing was performed by the UCLA Jonsson Comprehensive Cancer Center Genomic Shared Resource. A.H.L. is a pre-doctoral fellow supported by the UCLA Tumor Immunology Training Grant (USHHS Ruth L. Kirschstein Institutional National Research Service Award # T32 CA009120). L.S. was supported by a Career Enhancement Program award from the UCLA SPORE in Brain Cancer. J.C.K. received a grant from the Swiss Cancer Research foundation (project BIL KFS-4563-08- 2018) and the Kurt and Senta Herrmann foundation. W.H. was supported by grants from the NIH/NCI (1R01CA236910), the Melanoma Research Alliance (https://doi.org/10.48050/pc.gr.75700), the Margaret E. Early Medical Research Trust Grant, and the Parker Institute for Cancer Immunotherapy at UCLA. The schematic was created with BioRender.com.