Current Research Projects
Identification and Characterization of the Genetic Basis of the Corneal Dystrophies
Corneal dystrophies are a group of bilateral inherited disorders of the cornea that are either associated with the development of opacities in the normally clear cornea or endothelial dysfunction that may result in corneal edema and loss of corneal clarity. Several dystrophies that primarily affect the corneal endothelium have been described: Fuchs endothelial corneal dystrophy (FECD, MIM #610158), posterior polymorphous corneal dystrophy (PPCD, MIM #122000) and congenital hereditary endothelial dystrophy (CHED, MIM #217700). The prevalence of each of the corneal endothelial dystrophies varies significantly from one population to another, although they constitute common indications for corneal transplantation in published series from around the world.
The Cornea Genetics Laboratory has identified and reported dozens of novel mutations in genes associated with inherited disorders of the cornea and has identified the genetic basis of a number of different corneal dystrophies:
- Identification of the genetic basis of posterior amorphous corneal dystrophy
- Identification of the genetic basis of punctiform and polychromatic pre-Descemet corneal dystrophy
- Identification of the genetic basis of corneal dystrophy mapped to chromosome 10q23-q24 as epithelial recurrent erosion dystrophy
- The first to implicate the OVOL2 gene in the pathogenesis of PPCD1 and the first to confirm the association of truncating mutations in the ZEB1 gene with PPCD

Fig. 1. Slit-lamp photomicrographs of combined granular-lattice corneal dystrophy associated with the Met619Lys mutation in the TGFBI gene in various members of a single family, wherein the disease presents as opacifications in the cornea. (Aldave et al, Arch Ophthalmol. 2008;126(3):371-377).
The Cornea Genetics Laboratory is continuing to investigate the genetic basis of inherited corneal disorders such as Lisch corneal dystrophy and a recently identified corneal endothelial dystrophy with peripheral macular opacities using a variety of techniques, including whole genome sequencing, RNAseq expression analysis of affected corneal tissue and various functional assays.
Investigation of the Role of SLC4A11 in Corneal Endothelial Cell Biology and Disease
The solute linked carrier family 4 member 11 (SLC4A11) gene encodes a membrane protein that is highly expressed in the corneal endothelium, which consists of a monolayer of corneal endothelial cells that “pump” fluids across the inner layer of the cornea, maintaining the cornea in a slightly dehydrated state that is necessary for corneal clarity. Over 80 unique SLC4A11 mutations are reported to be associated with congenital hereditary endothelial dystrophy (CHED), a bilateral inherited disorder of the cornea that presents at birth or early childhood. SLC4A11 mutations are also associated with a subtype of Fuchs endothelial corneal dystrophy (FECD), which affects up to 5% of the world’s population above the age of 40, and is the most common indication for corneal transplantation in the US. Both CHED and FECD are characterized by the development of corneal endothelial dysfunction that results in corneal edema and impaired vision.
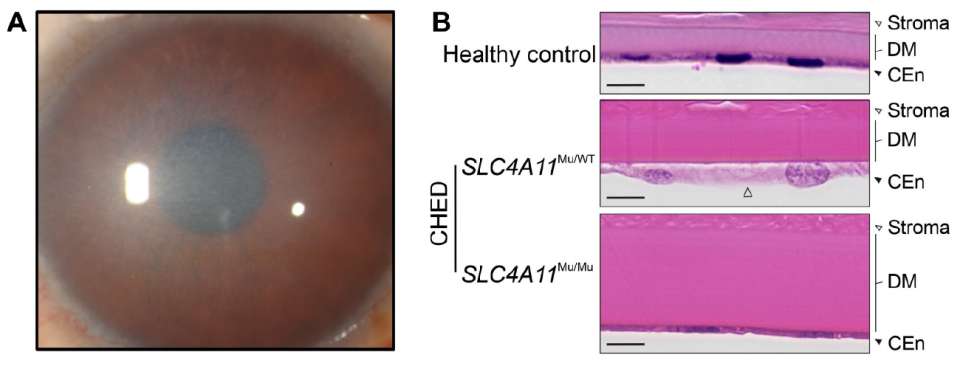
Fig. 2. (A) Characteristic diffuse corneal opacification in a pediatric patient affected with CHED. (B) Histopathologic examination of control and CHED corneas demonstrating Descemet membrane (DM) thickening and corneal endothelial cell (CEnC) alteration or attenuation in corneas from individuals with CHED (H&E stain). D, cytoplasmic inclusion. Scale bar: 10 μm. (Panel (B): Zhang et al, Invest Ophthalmol Vis Sci. 2020;61(8):39).
A. Transcriptomic characterization of the cellular changes induced by loss of SLC4A11
The Cornea Genetics Laboratory is using comparative transcriptomic approach to characterize the cellular
changes induced by loss of SLC4A11 in complementing cell-based (primary human corneal endothelial
cells, immortalized mouse and human corneal endothelial cells) and animal models (Slc4a11-/- mouse).
B. Considering sex as a biological variable in the pathogenesis of corneal endothelial diseases.
The Cornea Genetics Laboratory is investigating the sexual dimorphic response to loss of Slc4a11 at
phenotypic, transcriptomic, and functional levels using the Slc4a11-/- mouse model.
C. Characterization of the effect of SLC4A11 mutations on corneal endothelial cell biology and
SLC4A11 protein function.
To better understand the pathomechanisms via which SLC4A11 mutations lead to CHED or FECD, the
Cornea Genetics Laboratory is characterizing the impact of CHED- and FECD4- associated SLC4A11
mutations on corneal endothelial cell function (e,g. viability, cell barrier, migration) and SLC4A11 sub-
cellular localization and protein function using cell-based and molecular biology assays
D. Investigating extraocular phenotypes in Slc4a11-/- mouse.
The Cornea Genetics Laboratory is actively collaborating with principal investigators in other disciplines
to characterize the extraocular phenotypes induced by the loss of Slc4a11 in the Slc4a11-/- mouse model.
Preclinical Development and Characterization of AAV Gene Therapy to Treat Congenital Hereditary Endothelial Dystrophy
The Cornea Genetics Laboratory has been selected to participate in the Foundation for the NIH accelerating Medicines Partnership® (AMP®) Bespoke Gene Therapy consortium and received funding from the California Institute for Regeneration Medicine (CIRM) to perform preclinical studies of AAV gene therapy for CHED, an autosomal recessive, monogenic disorder associated with biallelic mutations in SLC4A11. These studies include performing efficacy, dose-optimization, toxicology, and biodistribution studies of AAV gene delivery of human SLC4A11 by intrastromal injection into the corneas of Slc4a11-/- mice, which are the only animal model of CHED. The outcomes of this study will form the basis for a Pre-IND filing and move AAV-SLC4A11 gene therapy for CHED closer to a first-in-human Phase I/II clinical trial.
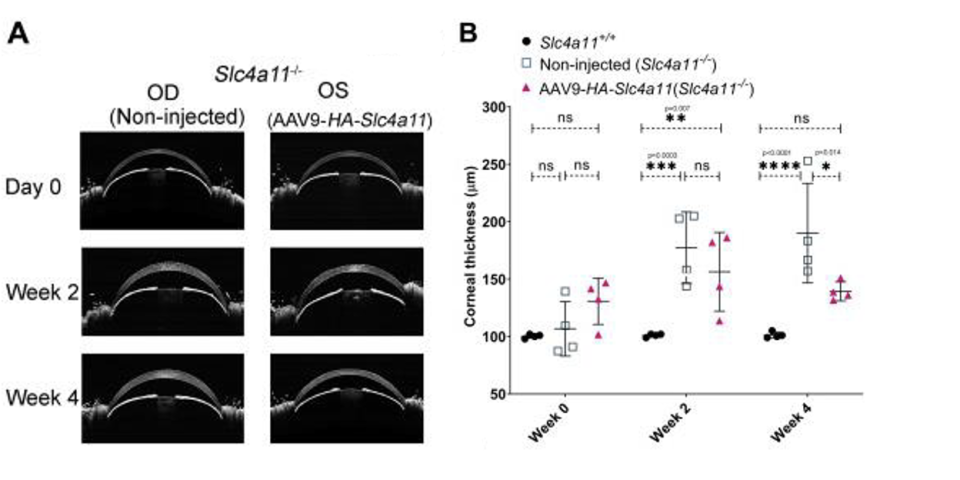
Fig. 3. Corneal edema is reduced with AAV9-mSlc4a11 in an animal model of CHED. (A) Representative OCT images of non-injected and AAV9-HA-Slc4a11 injected eyes of 5-week old Slc4a11-/- mice. (B) Quantification of data from panel (A). (Panels (A) and (B): Shyam et al, Ophthalmol Sci. 2022;2(1):100085.)
Characterization of Corneal Endothelial Cells During Ex Vivo Expansion
Corneal endothelial cell dysfunction is the primary indication for corneal transplantation in the United States and globally, accounting for 55% of all keratoplasty procedures performed in the US in 2018. However, the worldwide shortage of donor corneal tissue for the treatment of corneal endothelial dysfunction necessitates the development of viable alternatives to the current paradigm of one donor cornea restoring sight to only one recipient. While the prospect of CEnC expansion from ex vivo donor tissue brings hope for a new paradigm, where one donor cornea can restore sight to multiple recipients, significant challenges to the successful culture and expansion of CEnC in vitro exist, including cell state transition and cellular senescence. In an effort to overcome these challenges, the Lab is characterizing ex vivo corneal endothelial cells during cell culturing and expansion, and investigating the roles of oxidative stress and inflammation in mediating cell state transition and cell senescence in ex vivo corneal endothelial cells.
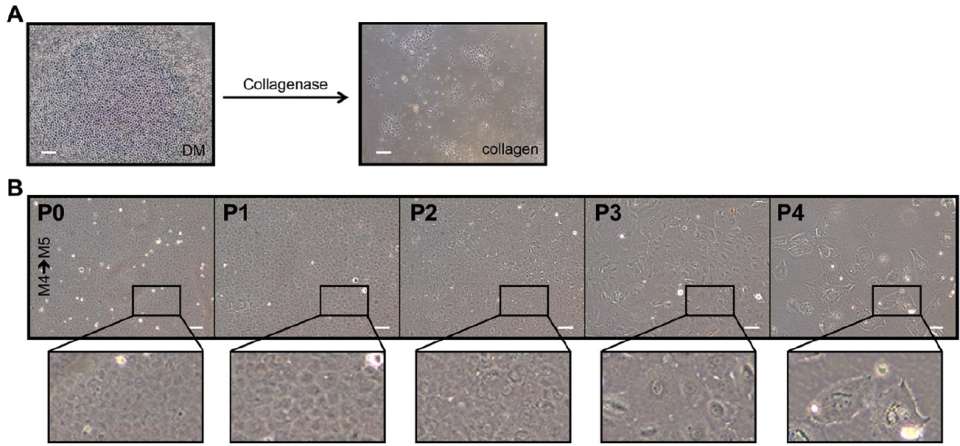
Fig. 4. Ex vivo corneal endothelial cells (CEnC) undergo cell state transition and cellular senescence during cell expansion. (A) CEnC dissociated from Descemet membrane using collagenase were seeded onto collagen-coated plastic. Images show cells 1 day after seeding. (B) Phase-contrast microscopy images show confluent CEnC cultured with M4/M5 media at five passages (P0-P4). Cell senescence and loss of CEnC morphology are typically observed at P3 and P4.