Brain Tumors
Find your care
Call 310-825-5111 to learn more about our world-class pediatric neurosurgery services.
Diagnoses
Medulloblastomas
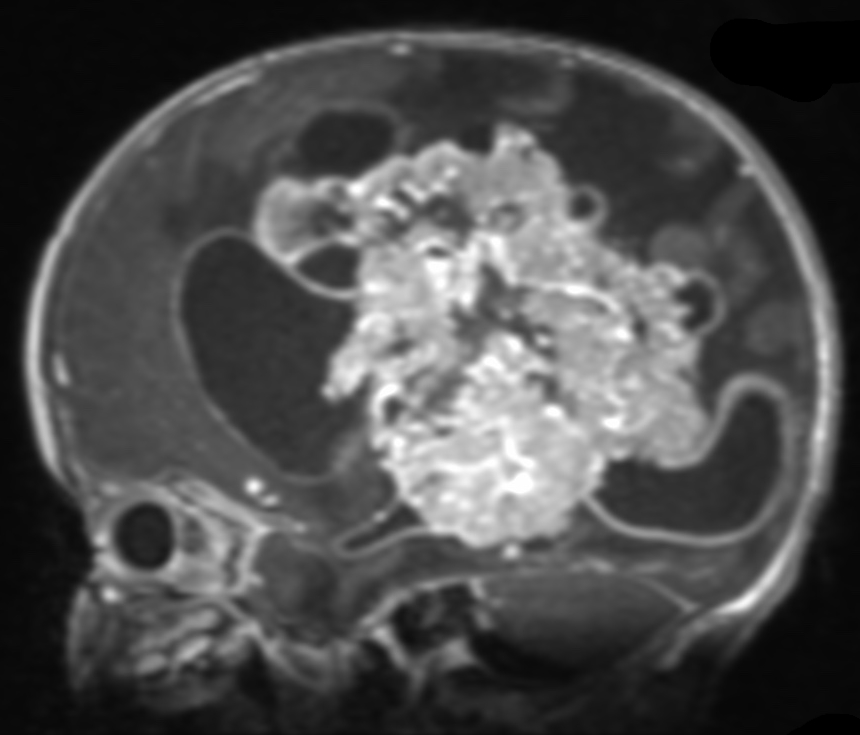
Medulloblastomas are the most common malignant brain tumors found in children. Between 250 and 500 children under the age of 16 are diagnosed with medulloblastoma each year in the United States, and it accounts for nearly 20% of all childhood brain tumors. Presenting symptoms are usually from obstructive hydrocephalus, and typically include headaches, nausea and vomitting, clumsiness, visual changes, and issues with coordination. Surgery is the first step in treating medulloblastoma, followed by chemotherapy and/or radiation therapy. The survival rates in children depend on the surgeon’s ability to safely and completely remove the tumor, and the sub-type of medulloblastoma. With optimal care, long-term survival rates can be better than 80%.
Pilocytic Astrocytomas

Pilocytic astrocytomas are another tumor type found most frequently in children in the cerebellum.
Symptoms arise as a result of increased intracranial pressure, and include headache, nausea and vomiting, clumsiness, visual changes, and issues with coordination. Pilocytic astrocytomas are relatively common, compromising about 20% of all brain tumors in children. The best treatment for these tumors is maximal safe surgical resection. With optimal care, long-term survival rates are well above 95%.
Atypical Teratoid/Rhabdoid Tumors
Atypical Teratoid/Rhabdoid Tumor (AT/RT) is a rare and fast growing malignant tumor of the brain and spinal cord. AT/RT is rare, found in fewer than 10% of brain tumors in children. It is also most prevalent in children aged 3 or younger, but a sub-set can also occur in older children. Presenting symptoms often include morning headache, vomiting, loss of balance and coordination, unusual fatigue, and increased head size in infants. Treatment involves a combination of surgery, chemotherapy, and radiation. Multiple forms of treatment are usually needed for AT/RT due to the severity of the tumor.
Ependymomas
Ependymomas in children most often occur in the posterior fossa, which includes the fourth ventricle, the brainstem, and the cerebellum. Ependymomas are relatively rare, and account for about 10% of all pediatric brain tumors. Symptoms vary, and are related to the location and the size of the tumor. The most important treatment is surgery, obtaining the maximal safe resection. The prognosis of this tumor is dependent on the extent of surgical resection, the presence or absence of disease spread, and the particular tumor sub-type.
Diffuse Midline Glioma with Histone H3-K27M Mutation
What was previously known as “DIPG” are malignant tumors that form within the brainstem in the central nervous system. Their rarity, location, and aggressive biology make these tumors extremely difficult to study and treat. Management of these lesions requires several difficult decisions. We typically recommend a surgical biopsy to establish the diagnosis, as we occasionally see a more benign tumor mimicking this appearance on imaging. Radiation therapy is the mainstay of treatment for children with malignant brainstem gliomas. In certain cases, chemotherapy and surgical resection may be options. We at UCLA are actively working in collaboration with the other major pediatric neuro-oncology centers around the world in developing new ways to treat this terrible disease.
Low Grade Gliomas
Supratentorial astrocytomas, oligodendrogliomas, gangliogliomas, and other low-grade gliomas are frequently undiscovered until early adulthood. Presenting symptoms can include headache, neurological deficits, or seizures, but these can often be found incidentally as well. Complete surgical resection is the most important form of treatment, with other treatment modalities typically reserved for cases of recurrence. Long-term survival rates exceed 90% for children diagnosed with low-grade gliomas, with optimal treatment.
Optic Pathway/Hypothalamic Gliomas
Optic pathway gliomas are low-grade astrocytic tumors that arise from within the optic nerves and hypothalami. They account for an estimated 5% of childhood brain tumors, and are almost always discovered in childhood. It has a particular association with type 1 neurofibromatosis. Due to the location, these tumors typically present by affecting vision and/or hormone function. Symptoms include worsening vision, stunted growth, or precocious puberty. Treatment for these patients should be provided by multi-disciplinary pediatric neuro-oncology programs, due to the complexity of their needs.
Craniopharyngiomas
Craniopharyngiomas are rare tumors that arise from cells near the pituitary gland. In the United States, there are only an estimated 350 cases of craniopharyngioma diagnosed each year. Craniopharyngiomas are mainly seen in children 5-14 years old, or in young adults. Adamantinomatous craniopharyngiomas are more common in the pediatric age group, whereas papillary craniopharyngiomas are predominately seen in adults. Craniopharyngiomas are most often benign, and become large before they are diagnosed. Symptoms include headaches, nausea, obesity, delayed development, impaired vision. Surgery to remove the tumor is usually the first step in treatment. Surgery can involve either an open craniotomy, or minimally-invasive endoscopic surgery; at UCLA, we offer extensive experience in either approach, and recommend one or the other on an individual basis. Radiation therapy can also be a treatment option in the event that not all of the tumor can be removed during surgery. In addition, an endocrinologist works with the treatment team to help develop a long-term care plan due to the proximity of the tumor to the pituitary gland and hypothalamus.
Primitive Neuroectodermal Tumors (PNET)
The group of tumors previously termed “PNET” are aggressive tumors that arise in the cerebral hemispheres. These tumors are relatively rare, and are most frequently seen in children younger than 5 years of age. Maximal safe surgical resection is typically the first step in treatment, but is administered in combination with intensive chemotherapy and high dose radiation therapy to the brain and spine. Much is still being learned about the individual diseases that make up this group of tumors.
Pineal Region Tumors
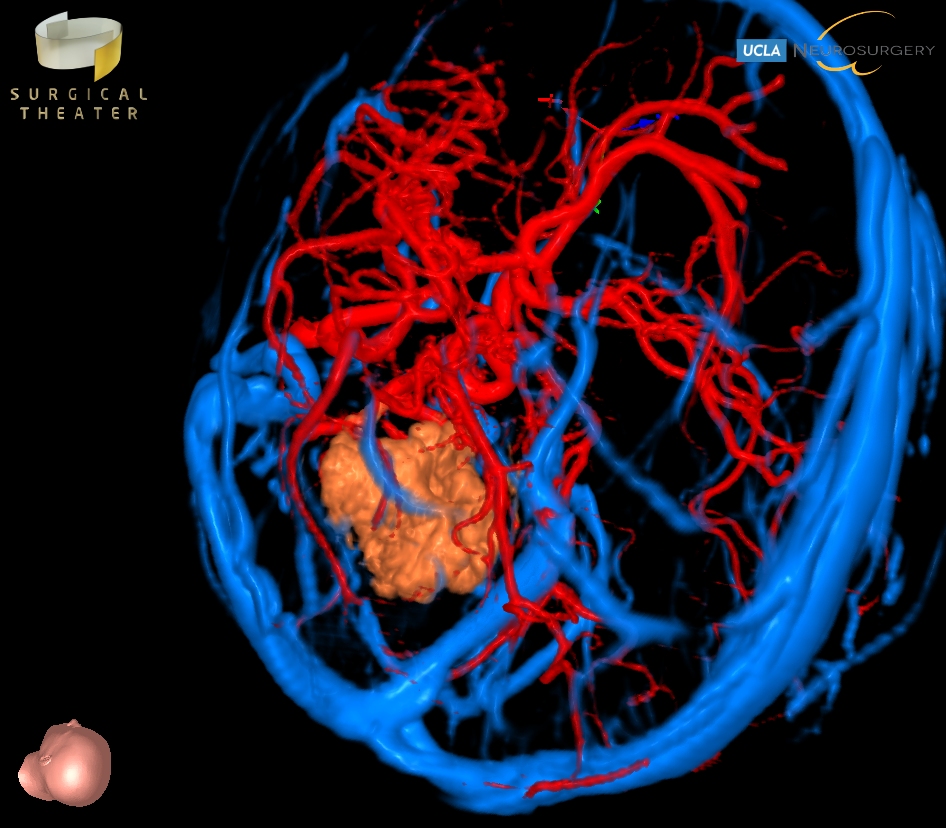
Pineal tumors arise in the pineal gland, which is in a deep area surrounding the midbrain. Pineal region tumors represent about 3-8% of childhood brain tumors, and include at least 17 different types of tumors that may occur in this region. The four most common types are germ cell tumors, pineal cell tumors, gliomas, and meningiomas. Most of these tumors are benign in biology. Symptoms associated with pineal region tumors are headache, nausea, memory disturbances, and visual changes, and hydrocephalus is often the primary cause for these symptoms. Treatments vary based on the particular tumor type, but when surgical resection is needed, this can be a particularly complex location, and requires an expert team. The prognosis for benign tumors in this region is excellent, with a high cure rate with optimal surgery.
Choroid Plexus Tumors
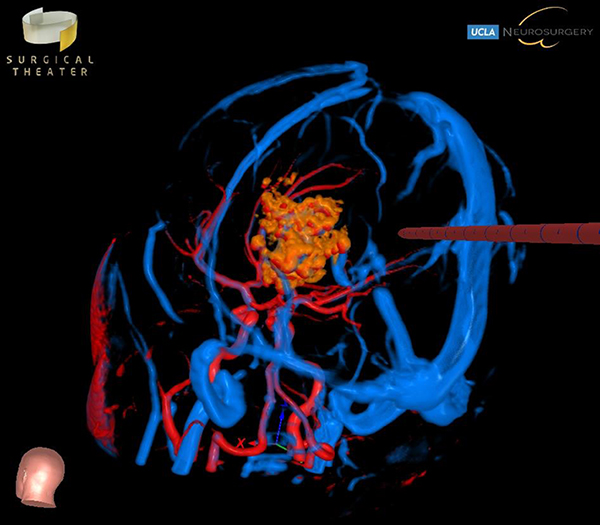
Choroid plexus tumors arise from the choroid plexus within the ventricles of the brain. These tumors are rare, representing only about 3% of brain tumors in children. About 10-20% of brain tumors found in the first year of life are choroid plexus tumors. The majority of choroid plexus tumors are either choroid plexus papillomas (CPP) or choroid plexus carcinomas (CPC). CPPs are benign and far more common, accounting for 80-90% of choroid plexus tumors. CPCs are malignant, and are more likely to spread to other parts of the brain and spinal cord. Symptoms for these tumors include headaches, nausea, lethargy, irritability, problems with development, and enlarged fontanelles. All of these are a direct result of the increased intracranial pressure and of the hydrocephalus that may be caused by the tumor. Surgical resection is the mainstay of treatment, but can be particularly complex in these cases, and should be performed by an experienced pediatric neurosurgery team.