Precision therapies targeting melanoma and other cancers have improved patient survival, but tumors often stop responding to drugs, and recent studies point to changes in DNA – both inside and outside chromosomes – as a driving factor in tumor genesis as well as in treatment resistance.
To investigate mutations in a key cancer-promoting genetic pathway, UCLA Jonsson Comprehensive Cancer Center researchers led the development of a melanoma model of drug resistance, enabling them to study structures and dynamics resulting in intrachromosomal and extrachromosomal changes that support resistance in cancer cells. The model also allowed them to experimentally manipulate drug dose regimens and observe resistance-related responses, possibly leading to therapeutic approaches that will extend effectiveness.
“Tumor genomes, especially those of aggressive cancers, have an ability to change rapidly and unpredictably. With the numerous advances in genomic sequencing, mapping and data analysis technologies, we are beginning to understand conditions, structures and interactions at the chromosomal level that influence oncogenesis and drug resistance. In this study, we describe mechanisms involved in focal amplification of BRAF and other genes in the MAPK signaling pathway, which is a driver of many cancer types,” said Thomas Graeber, PhD, a researcher with the UCLA Jonsson Comprehensive Cancer Center and director of the UCLA Metabolomics Center, senior author of an article published online Dec. 20 at Cancer Discovery (DOI: 10.1158/2159-8290.CD-20-0936).
BRAF mutations, relatively common in thyroid cancer and melanoma, have been targeted using drugs such as vemurafenib and dabrafenib and other MAPK pathway inhibitors. Resistance often occurs, however, through reactivation of this pathway, activation of a different pathway, or both. One mechanism of MAPK pathway reactivation often seen in melanoma is the acquisition of BRAF amplifications.
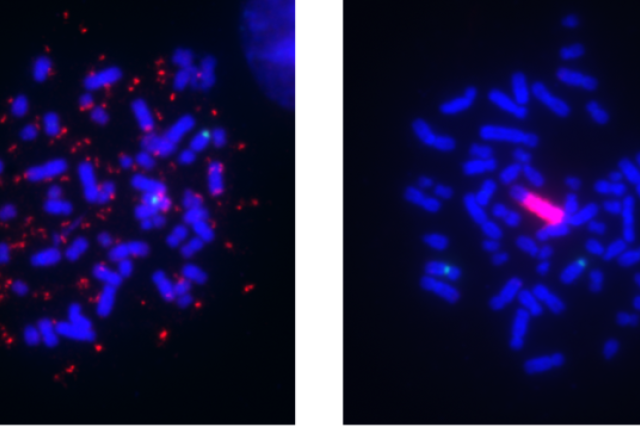
A segment of chromosomal DNA that undergoes amplification, known as an amplicon, can lead to overexpression of cancer-promoting genes and those that interfere with targeted therapies. However, unlike DNA in normal human cells, this DNA can be separate from the chromosomes and thus more freely floating in the nucleus. This type of amplicon is known as extrachromosomal DNA (ecDNA), originally termed double minutes (DMs) due to how they appear under the microscope.
With time, these amplicons can reintegrate into a different chromosome, and these sites of integration are known as homogeneously staining regions (HSRs) because they contain long stretches of amplicon copies aligned in a row. Both of these forms of focal amplifications are known contributors to cancer development and drug resistance, and the switch between the DM and HSR mode is an example of how cancer cells can evolve, or exhibit ‘plasticity.’ Although they were first observed more than 60 years ago, only within the last few years have technological advances begun to fully reveal their structure, organization and role in tumor evolution.
“This study was designed to provide details about the structure, dynamics and vulnerabilities of MAPK-related focal amplifications as they relate to drug resistance in melanoma,” said Kai Song, a graduate student in the Graeber Lab and the article’s first author. “A complete knowledge of these structures and interactions is central to our understanding of tumor evolution and plasticity and to the development of therapeutic approaches that overcome resistance.”
The researchers treated a human melanoma cell line with kinase inhibitors that target the MAPK signaling pathway to develop a model of drug resistance. Initially, high BRAF gene amplification was seen in the extrachromosomal DNA form, but over the course of months in culture at a stable drug dose, the mechanism switched to the intrachromosomal HSR form. According to the authors, the observance of a mode switch in melanoma adds to several related reports in the literature of similar events in focally amplified oncogenes of different cancer types, including leukemia and neuroblastoma.
The study found that each form of DNA offered different ‘fitness’ advantages to tumor cells, such as in response to changing therapeutic conditions. For example, when cells are exposed to constant, steady drug dose treatment, intrachromosomal HSRs appear to be “more fit” than their extrachromosomal counterparts to promote cancer growth – supporting a fitness-driven evolution from DMs to HSRs in these conditions. However, a non-steady or oscillating drug dose challenge creates an advantage for not having the amplicon copy number tied to chromosomes, and thus DM-harboring cells are retained.
Even though DM-harboring cells are traditionally thought to be more plastic than HSR cells, the research found that HSR cells also possess amplicon copy number plasticity. HSR length can shorten when drug dose is reduced, allowing the cancer cells to escape the “drug addiction” that comes along when they gain drug resistance. The mechanism by which HSR shortening occurs, removal of amplicons without further changes in the HSR, was revealed through DNA structural analysis conducted in collaboration with a team at the Computer Science Department of the University of California, San Diego, under the direction of Vineet Bafna, PhD.
The findings were bolstered by reports in the literature and clinical studies from a team led by collaborator Dr. Roger Lo, a member of the UCLA Jonsson Comprehensive Cancer Center. They find evidence for the plasticity of focal amplifications of MAPK genes in patient tumors and in patient-derived xenograft-model tumors that have become resistant to MAPK inhibitor treatments.
Among other significant results, the research team documented that although melanoma cells may become resistant to signaling pathway-inhibiting drugs, treatment with the drugs appears to make them vulnerable to a different form of treatment. With MAPK inhibition, melanoma cells shift their major energy generation program from glycolysis, or glucose metabolism, to mitochondrial pathways in which respiration and energy production occur within the cells. This leads to elevated production of reactive oxidative stress in the treated cells. This cascade has been observed in previous studies, and the UCLA-led research team extended the findings in the focal amplification context to identify the involvement of oxidation of fat molecules in the cell. This insight revealed that the oxidatively stressed cells are vulnerable to a new class of drugs, so-called pro-ferroptotic drugs, which trigger oxidized lipid-mediated programmed cell death.
“An important value of this model system is that it is allowing us to hunt for new vulnerabilities of cancer cells that use these focal amplification-based methods of resistance. Therapeutically targeting these vulnerabilities is our end goal,” Graeber said.
Article: Plasticity of extrachromosomal and intrachromosomal BRAF amplifications in overcoming targeted therapy dosage challenges; Published online Dec. 20 at Cancer Discovery (DOI: 10.1158/2159-8290.CD-20-0936).




