Genetic Syndromes
Find Your Care
We deliver effective, minimally invasive treatments in a caring environment. Call 310-267-7838 to connect with an expert in endocrine surgery.
Genetic Syndromes / Rare Endocrine Diseases
Genetic syndromes are medical conditions caused by inherited mutations in our genes. In other words, these can be passed down from parent to child and run in families. These are very rare diseases that occur in about 1 in every 500,000 or 1,000,000 cases. Due to our large volume of patients, we collectively built expertise in managing patients with the following genetic syndromes that affect our endocrine organs.
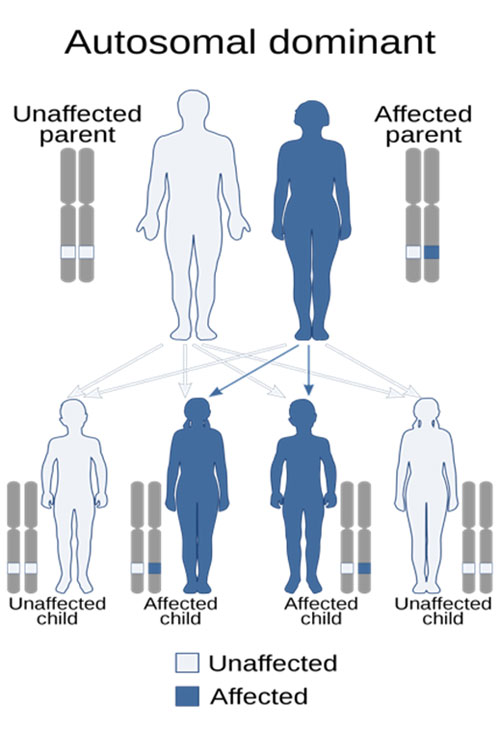
When Do Patients with Thyroid, Parathyroid, and Adrenal Diseases Need Genetic Testing?
The vast majority of thyroid and parathyroid diseases do not have a genetic component. Every patient seen for thyroid and parathyroid disease is asked about their family history. When there is a clear pattern of disease across family members, or certain subtypes of thyroid or parathyroid disease, we will refer patients to our geneticists.
In contrast, approximately 1/3 of pheochromocytoma cases are now known to be genetic, which means that they have a hereditary or familial association. The most common associated genetic conditions are Multiple Endocrine Neoplasia (MEN) type 2 and von Hippel-Lindau Syndrome, followed by Neurofibromatosis type 1 and Familial Paraganglioma Syndrome (which is caused by mutations in the succinate dehydrogenase gene). We recommend genetic counseling for all patients with pheochromocytoma or paraganglioma, as this can provide important information for the patient’s prognosis and earlier screening for family members.
For more information on genetic counseling and testing at UCLA >
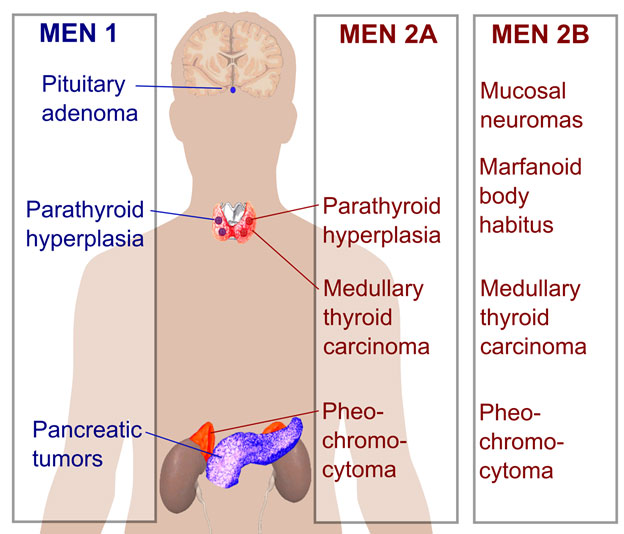
What are Multiple Endocrine Neoplasia (MEN) syndromes?
Multiple Endocrine Neoplasia (MEN) syndromes are passed down through families. A mutated gene causes different endocrine glands in the body to develop benign and cancerous neuroendocrine tumors. Endocrine glands secrete hormones, so tumors arising from these glands may also overproduce hormones that result in symptoms. Early diagnosis and treatment are essential to help reduce the impact of these conditions on the patient.
There are several forms of MEN syndromes:
- MEN-I
- MEN-IIA
- MEN-IIB
- Familial medullary thyroid cancer (variant of MEN-IIA)
- MEN-IV
What is Multiple Endocrine Neoplasia Type 1 (MEN-I)?
Multiple Endocrine Neoplasia (MEN) I is a hereditary disorder in which one or more of the following glands develop excess normal tissue (hyperplasia) or adenoma (tumor): the parathyroid, the pancreas, the pituitary, and (rarely) the adrenals and thyroid gland. These are all endocrine glands, which produce and secrete hormones into the blood or lymph systems.
What causes MEN-I?
The cause of MEN I is genetic. Tumors of various glands appear in the same person, but not necessarily at the same time. The disorder is inherited, may occur at any age, and affects men and women equally.
What are the signs and symptoms of MEN-I?
Most people affected with this syndrome seek medical treatment because of one of the following: peptic ulcer disease, symptoms related to low blood sugar, symptoms related to high serum calcium levels or kidney stones, or symptoms related to pituitary problems (such as headache).
Risk factors are a family history of this disorder, a previous pituitary tumor, and a history of Zollinger-Ellison syndrome.
Symptoms:
Symptoms vary greatly from person to person and may be related to peptic ulcer disease, hypoglycemia, hypercalcemia, or pituitary dysfunction.
- Abdominal pain
- Burning, aching, or hunger discomfort in the upper abdomen or lower chest that is relieved by antacids, milk, or food
- Black, tarry stools
- Nausea and vomiting
- Bloated feeling after meals
- Weakness
- Headache
- Vision problems
- Loss of coordination
- Anxiety
- Mental changes or confusion
- Coma if hypoglycemia is untreated
- Loss of appetite
- Muscle pain
- Fatigue
- Sensitivity to the cold
- Unintentional weight loss
- Low blood pressure
- Loss of axillary or pubic hair
- In women, cessation of menses, infertility, or failure to lactate
- In men, decreased sexual interest, loss of body or facial hair
What treatments are needed for patients with MEN-I?
Treatment depends on which endocrine glands are affected. Surgical removal of the affected gland is the treatment of choice when feasible. For pituitary adenomas, a medication called bromocriptine may be used instead of surgery in select cases. Hormonal replacement therapy is indicated when glands are removed or secretion is inadequate.
What is the prognosis for patients with MEN-I?
Pituitary and parathyroid tumors are usually benign, but some tumors may become malignant (cancerous), accounting for overall decreased life expectancy. The symptoms of peptic ulcer disease, hypoglycemia, hypercalcemia, or pituitary dysfunction should respond to treatment.
What is Multiple Endocrine Neoplasia (MEN) Type II?
Multiple Endocrine Neoplasia II (MEN II) is a hereditary disorder in which a type of thyroid cancer accompanied by recurring cancer of the adrenal glands.
What causes MEN-II?
The cause of MEN II is genetic -- a mutation in a gene called RET. Multiple tumors may appear in the same person, but not necessarily at the same time. The adrenal tumor is a pheochromocytoma and the thyroid tumor is a medullary carcinoma of the thyroid. The disorder may occur at any age, and affects men and women equally. The main risk factor is a family history of MEN II.
What is the difference between MEN-IIA and MEN-IIB?
MEN-IIA can cause medullary thyroid cancer, hyperparathyroidism, and pheochromocytoma.
MEN-IIB can cause medullary thyroid cancer, pheochromocytoma, and mucosal neuromas (bumps on the lips and tongue).
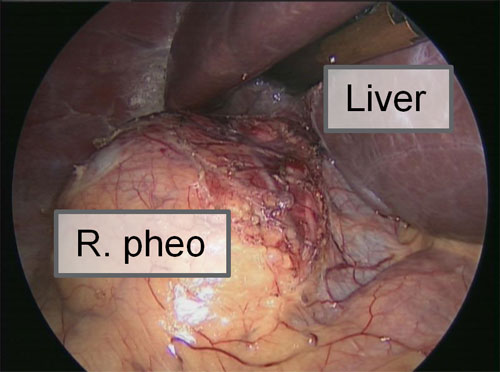
What are the signs and symptoms of MEN-II?
- Severe headache
- Heart palpitations
- Rapid heart rate
- Sweating
- Chest pain
- Abdominal pain
- Nervousness
- Irritability
- Loss of weight
- Diarrhea
- Cough
- Cough with blood
- Fatigue
- Back pain
- Increased urine output
- Increased thirst
- Loss of appetite
- Nausea
- Muscular weakness
- Depression
- Personality changes
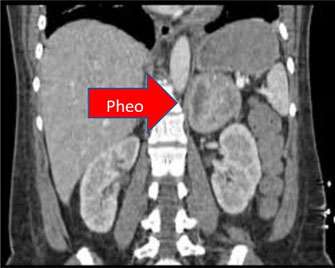
The symptoms may vary, but are consistent with those of pheochromocytoma, medullary carcinoma of the thyroid, and sometimes hyperparathyroidism.
What treatments are needed for patients with MEN-II?
Surgery is needed to remove both the medullary carcinoma of the thyroid and the pheochromocytoma. Medullary carcinoma of the thyroid must be treated with total removal of the thyroid gland and removal of surrounding lymph nodes. Hormone replacement therapy is given after surgery.
Family members should be screened for the RET gene mutation.
What is the prognosis for patients with MEN-II?
Pheochromocytoma is usually benign (not cancer), but the accompanying medullary carcinoma of the thyroid that characterizes this condition is a very aggressive and potentially fatal cancer. Nonetheless, early diagnosis and surgery can often lead to cure.
What is MEN-IV?
Multiple endocrine neoplasia type 4 (MEN4) is a very rare form of MEN, an inherited cancer syndrome, characterized by parathyroid and anterior pituitary tumors, possibly associated with adrenal, renal, and reproductive organ tumors.
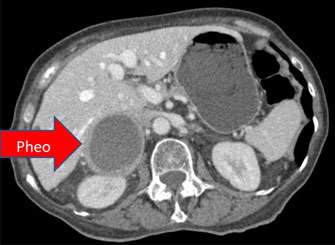
What are Succinate Dehydrogenase (SDH) syndromes?
SDH is a gene that help suppress the growth of tumors. In individuals that have a mutation in this gene are more prone to paragangliomas and pheochromocytomas, though it does not occur in everyone. These tumors can cause symptoms by increasing in size or by over secreting “fight or flight” hormones, causing episodes of high blood pressure, anxiety, and headache.
What is Von Hippel-Lindau Syndrome?
This syndrome is caused by a mutation in the VHL gene that helps prevent the growth of tumors. Approximately 10-34% of patients with VHL syndrome may develop pheochromocytomas or paragangliomas, in addition to hemoangioblastomas, retinal angiomas, renal cell cancers, and neuroendocrine tumors of the pancreas.
What is Neurofibromatosis type I?
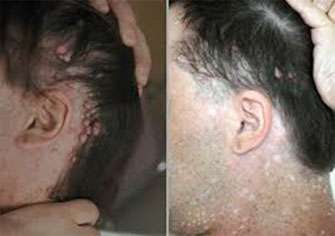
Neurofibromatosis type I results from a mutation in the NF1 gene, which similar to many of the genes above, prevents cells from growing too quickly. Without this gene, individuals develop many benign tumors of the nerves and skin and abnormal areas of pigmentation on their skin. Patients with neurofibromatosis type I can have cognitive abnormalities and are at risk for certain cancers, including soft tissue sarcomas. Up to 6% of patients with NF-1 also develop pheochromocytomas and paragangliomas.