Pediatric Craniosynostosis Program
Find your care
Call 310-825-5111 to learn more about our world-class pediatric neurosurgery services.
The craniosynostosis program at UCLA has a long tradition of excellence and is responsible for training many of the current leaders in the field. Since the inception of the multi-disciplinary craniofacial clinic in 1972, we have defined the standard of care for the treatment of craniofacial abnormalities. We have an impeccable track-record of superior cognitive and cosmetic outcomes with no mortalities or serious infections.
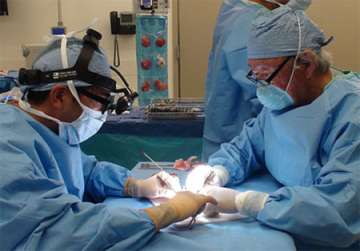
Our Pediatric Neurosurgeons both have extensive fellowship-training and expertise from the most common to the most rare types of craniofacial surgery. Similarly, the Craniofacial Surgeons in the UCLA Division of Plastic & Reconstructive Surgery are all board-certified in plastic surgery and fellowship trained in craniofacial surgery—all three of them completed their fellowship training at UCLA and were trained by Henry K. Kawamoto, MD, DDS, one of the ground-breaking surgeons who helped create and refine many of the surgical techniques used to correct craniofacial deformities.
Our surgeons work in close collaboration with the UCLA Craniofacial Clinic to develop a comprehensive and individualized treatment plan for each of our patients. The clinic utilizes a team approach to provide patients with individualized care. The clinic brings together neurosurgeons, plastic surgeons, oral surgeons, head and neck surgeons, medical geneticists, ophthalmologists, orthodontists, otolaryngologists, pediatricians, audiologists and speech pathologists to develop a comprehensive and coordinated treatment plan for each patient. The program also serves as a regional center for the southwestern United States.
Pediatric Neurosurgeons


Pediatric Craniofacial surgeons



The most common indications for craniofacial surgery are:
- Apert syndrome
- Congenital and Acquired Skull Defects
- Craniosynostosis
- Crouzon disease
- Facial and nasal encephaloceles with associated craniofacial disorders
- Pfeiffer syndrome
- Saethre Chotzen syndrome
Our pediatric neurosurgeons and pediatric craniofacial surgeons also extend their reconstructive expertise to patients of all ages with trauma-related injuries requiring craniofacial restoration and to pediatric patients requiring facial reconstruction following cancer surgery.
What is Craniosynostosis?
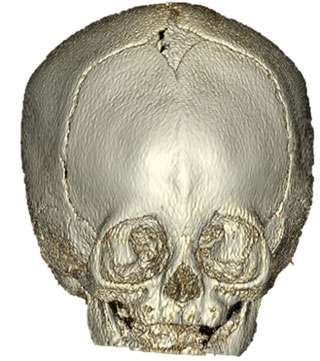
Craniosynostosis is the premature closure or one or more gaps between the growing bones of the skull. This condition may be present at birth or may be discovered shortly after birth. For some babies, this can be more subtle and an experienced craniofacial plastic surgeon and/or a pediatric neurosurgeon is required to make the diagnosis. The most common form involves the sagittal suture (sagittal craniosynostosis), and is characterized by a long, narrow head (scaphocephaly or dolichocephaly), with ridging over the sagittal suture. Less common are coronal craniosynostosis, characterized by asymmetry of the forehead and orbits, and metopic craniosynostosis, in which there is a triangular appearance to the forehead with ridging in the middle of the forehead over the metopic suture.
Is it important to differentiate between positional plagiocephaly and craniosynostosis? Yes. Positional plagiocephaly is a flat spot typically on the back of an infant’s head caused by prolonged pressure on the bones of the skull before or after birth. This is a condition that is commonly self-limited and may be observed or treated with a helmet. It is important to differentiate this from craniosynostosis as the treatment of positional plagiocephaly is rarely surgical.
How common is Craniosynostosis?
Craniosynostosis is estimated to affect one out of every 2500 births.
Are there genetic links to Craniosynostosis?
Several genetic disorders, including Crouzon Syndrome and Apert Syndrome are associated with craniosynostosis. In addition, mutations of several genes have been identified in certain forms of craniosynostosis. A genetic consult can be valuable in not only diagnosis the syndrome but can be helpful to estimate the chances of having another infant with craniosynostosis for each family. Also, several non-genetic factors have been implicated in the origins of craniosynostosis, including fertility treatments, paternal profession and such environmental exposures are maternal smoking and certain drugs.
Do you perform minimally invasive endoscopic surgery for craniosynostosis?
Yes. We perform minimally invasive endoscopically assisted surgery for selected infants with sagittal or metopic synostosis and this is supplemented by the use of helmets after surgery. The children have much smaller scalp incisions, experience less blood loss and tolerate this surgery exceptionally well. This option is used especially in very young infants. Not all infants may be a good candidate for this procedure.
What are several common types of Craniosynostosis?
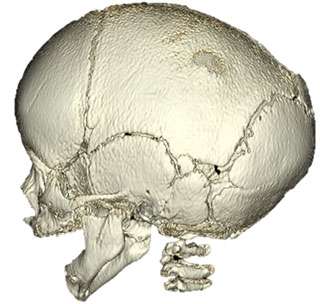
Sagittal Craniosynostosis
Sagittal synostosis is the premature closure of the sagittal suture that is located on the top of the head, running from front to back. A ridge on the top of the head can usually be felt through the scalp. When this suture closes early, the baby begins to have an elongation of the head from front to back (scaphocephaly) with narrowing of the temple region (bitemporal narrowing). Some babies will develop a prominent forehead (also called frontal bossing).
Unilateral Coronal Synostosis
There are two coronal sutures, each running from the top of the head down the sides in front of the ears. When one of these sutures closes prematurely, the baby begins to develop flatness of the forehead on the affected side. A ridge over the affected suture may be felt through the scalp. The eyebrow may appear raised on the affected side. The ear on the affected side will appear more forward when looking from the top (bird’s eye view). The top of the nose or nasal bridge will be deviated toward the affected side while the tip of the nose will point toward the unaffected side. The chin will appear slanted towards the affected side.
Bilateral Coronal Synostosis
When both coronal sutures are affected, a ridge can be felt on both sides of the head running from the top of the skull down the sides in front of the ears. Depending how early this is discovered, the forehead will appear flat. This will, in turn, make the eyes appear as if they are sticking out. The head will appear taller and wider than usual.
Lambdoidal Synostosis
There are two lambdoidal sutures in the skull, which meet in the back of the head in an upside down configuration. This is the rarest form of craniosynostosis and comprises only one percent of all cases. It is important to differentiate this condition from positional plagiocephaly which is quite common and typically does not require surgical correction.
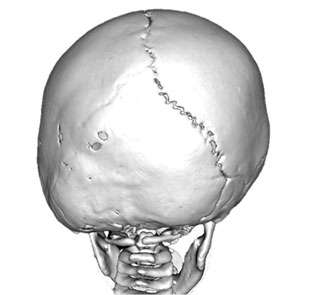
Is surgery necessary for Craniosynostosis?
Most cases require early surgery to prevent distortion of the face and skull. In addition, in certain instances, there may be evidence of increased pressure within the skull that may be associated with abnormal brain development. Failure to treat this may be associated with worse cognitive development. Some mild cases of craniosynostosis may not require surgery.
How dangerous is surgery for Craniosynostosis?
At UCLA, there have been no mortalities or serious infections after surgical procedures for craniosynostosis, attesting not only to the expertise of the surgeons, but of the entire team, including the pediatric anesthesiologists. Except for the most complex craniofacial syndromic cases, children are generally are discharged within 72 hours of surgery.